


")
")
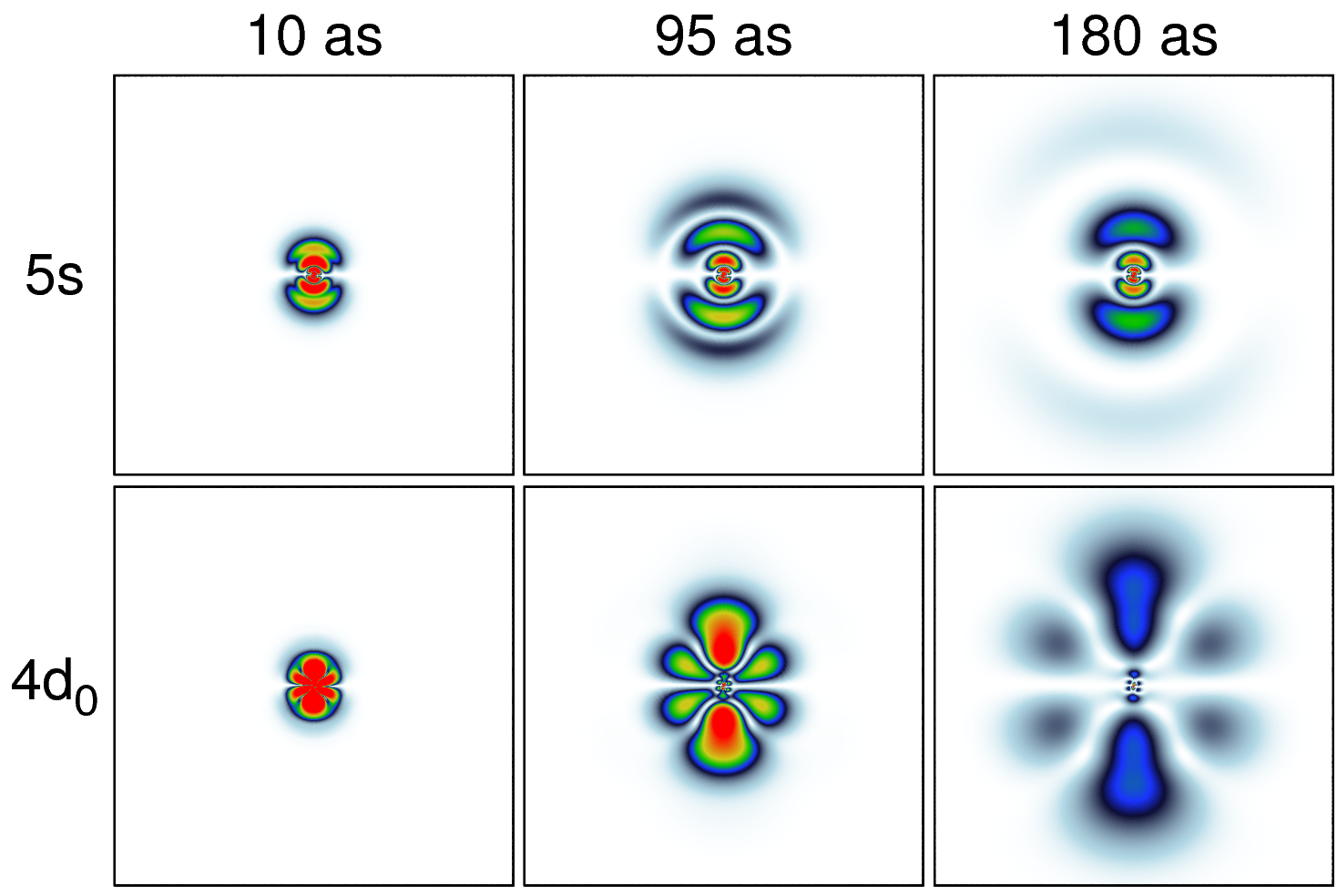
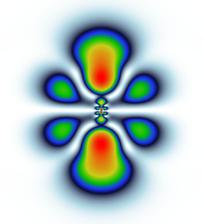
Figure 1: Probability densities of the photoelectron originating from the 4d0 and 5s shells of xenon are shown for different times after the ionizing attosecond pulse. The pulse has a duration of 10 as, a photon energy of 136 eV, and a peak electric field strength of 25 GV/m.
Stefan Pabst1,2, Loren Greenman3, Phay J. Ho4, David A. Mazziotti3, and Robin Santra1,2,*
1. Center for Free-Electron Laser Science, DESY, Notkestrasse 85, 22607 Hamburg, Germany
2. Department of Physics, University of Hamburg, Jungiusstrasse 9, 20355 Hamburg, Germany
3. Department of Chemistry and The James Franck Institute, The University of Chicago, Chicago, Illinois 60637, USA
4. Argonne National Laboratory, Argonne, Illinois 60439, USA
Published as: "Decoherence in attosecond photoionization", Phys. Rev. Lett. 106, 053003 (2011)
Attosecond science holds the promise of controlling electron motion to manipulate physical processes at the atomic level. One way of inducing electron motion is photoionization using an attosecond laser pulse. The typical time scale of electronic motion in atoms, molecules, and condensed matter systems ranges from a few attoseconds (1as = 10 -18s) to tens of femtoseconds (1fs = 10 -15s) [1]. Remarkable progress in high-harmonic generation made it possible to generate attosecond pulses as short as 80 as [2]. Attosecond pulses have opened the door to real-time observations of the most fundamental processes on the atomic scale like photoionization [3], Auger decay [4], and valence electron motion driven by relativistic spin-orbit coupling [5]. The focus of our paper is to answer an open fundamental question about electron control via attosecond photoionization: Can the nonstationary state of the parent ion be described by a Schroedinger wave function, i.e., is the state coherent?
Pulses with sufficiently broad coherent bandwidth can now bridge the energy splitting between valence and inner-shell atomic orbitals. One might expect that by ionizing these orbitals using an attosecond pulse, a coherent superposition of the corresponding ionic eigenstates is formed. While the entire system---ion plus photoelectron---is described by a wave function, the ion alone however must be described by a density matrix. This opens up the possibility that the state of the ion is not perfectly coherent. We show that the Coulomb interaction between the photoelectron and the parent ion triggers complex many-body effects, which unexpectedly enhance the entanglement between photoelectron and ion---leading subsequently to decoherence within the ion. We point out strategies for controlling the decoherence, offering new opportunities for x-ray free-electron lasers.
In this Letter, we analyze the creation of hole states via single-photon ionization in atoms using a single extreme-ultraviolet attosecond pulse (see Fig. 1). We investigate the creation of hole states via attosecond photoionization using the implementation of the time-dependent configuration-interaction singles (TDCIS) approach described in Ref. [6]. TDCIS allows us to study ionization dynamics beyond the common single-channel approximation and to understand systematically the relevance of multiple channels in the hole creation process.
Furthermore, we study the impact of the freed photoelectron on the remaining ion and demonstrate that the interaction between the photoelectron and the ion cannot be neglected for currently available state-of-the-art attosecond pulses. In particular, the interchannel coupling of the initially coherently excited hole states greatly enhances the entanglement between the photoelectron and the ionic states. Interchannel coupling is mediated by the photoelectron and mixes different ionization channels, i.e., hole configurations, with each other. Consequently, the degree of coherence among the ionic states is strongly reduced, making it impossible to describe the subsequent electronic motion in the ion with a pure quantum mechanical state.
The degree of coherence can be expressed in terms of entries of the reduced ion density matrix 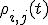 [6]---an observable quantity, as successfully demonstrated in Ref. [5]. The degree of coherence between ionic eigenstates is defined as
[6]---an observable quantity, as successfully demonstrated in Ref. [5]. The degree of coherence between ionic eigenstates is defined as 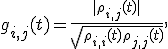 where
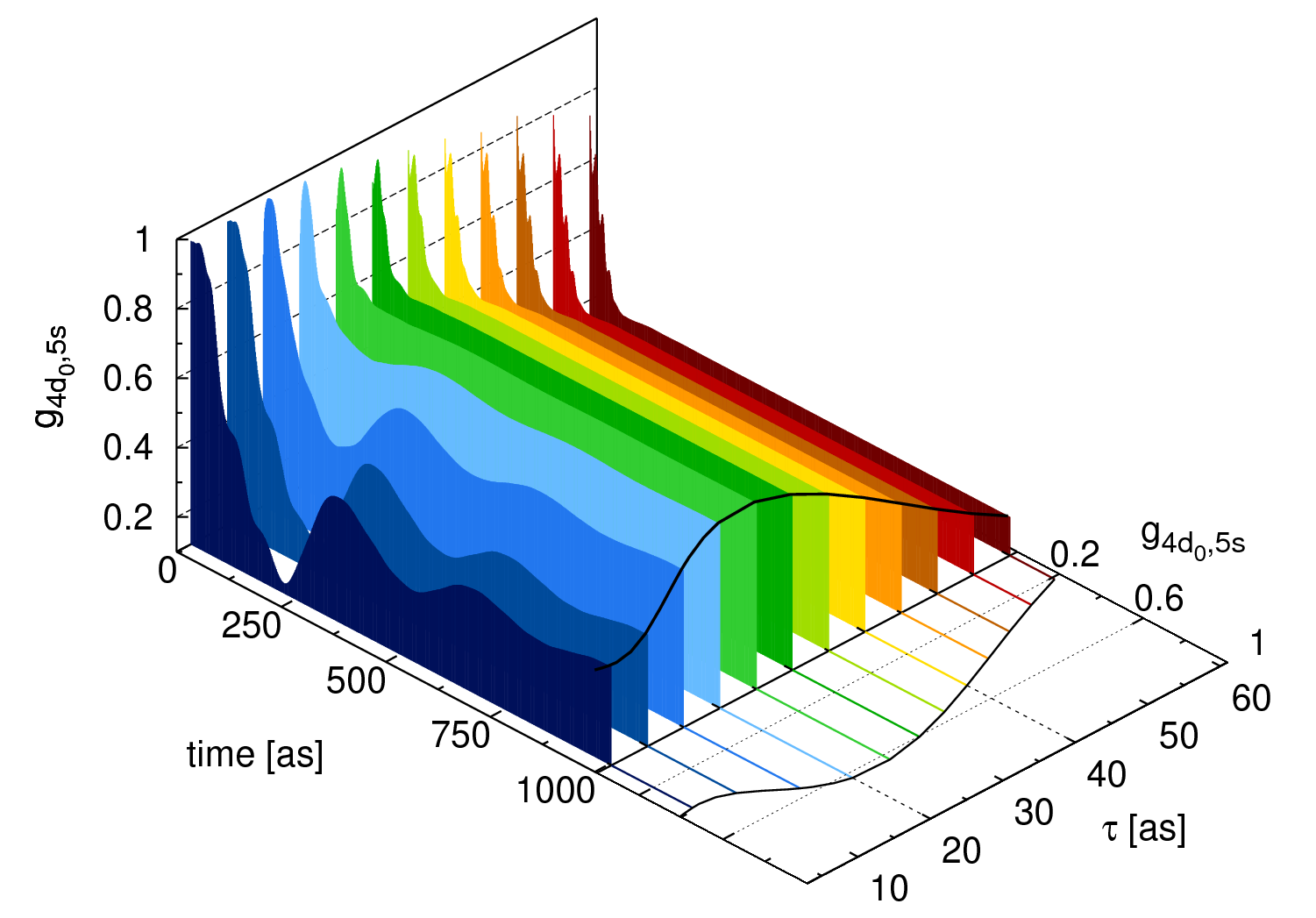
where 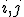 are indices corresponding to a pair of orbitals occupied in the neutral atom. Our particular focus lies here on the coherence between the 4d0 and 5s hole states of xenon (see Fig. 2).
are indices corresponding to a pair of orbitals occupied in the neutral atom. Our particular focus lies here on the coherence between the 4d0 and 5s hole states of xenon (see Fig. 2).
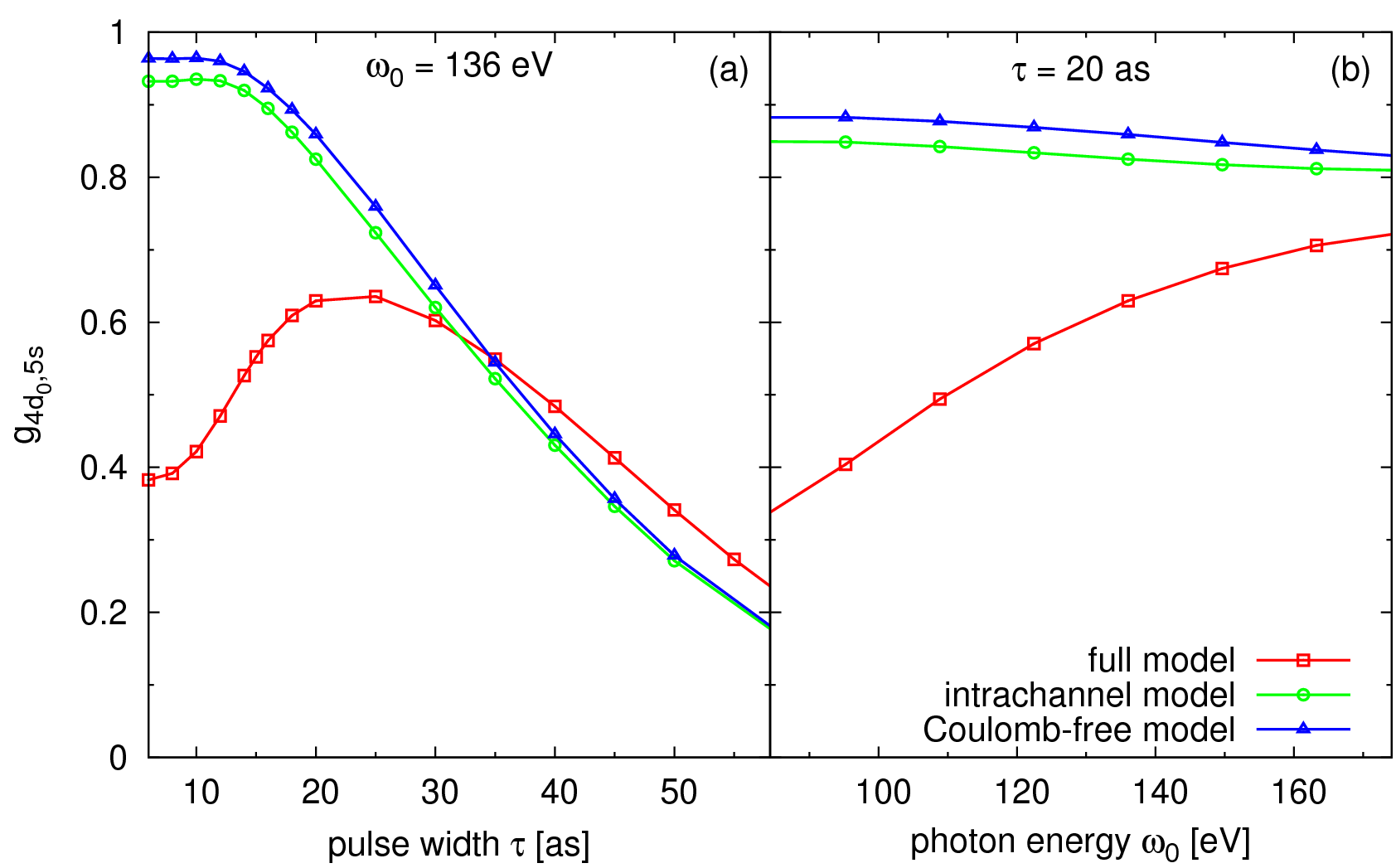
In Fig. 3 we investigate different Coulomb approximations and analyze their impact on  . By ignoring the residual Coulomb interaction between the photoelectron and the ion (blue curve), the trends seen in Fig. 3(a) can be intuitively understood by comparing the spectral bandwidth of the attosecond pulse with the energy splitting between the ionic states. By including Coulomb interactions within a given channel (green curve), the trends shown in Fig. 3 do not differ qualitatively from the first case, where all Coulomb interactions are ignored. In this second model, the excited electron can only interact with the occupied orbital from which it originates. Interactions between different occupied orbitals are considered only in the full model (red curve). We can clearly see that by including these interchannel coupling terms the degree of coherence differs dramatically for short attosecond pulses (see Fig. 3(a)) and, surprisingly, starts to drop due to an enhanced interaction between the slow photoelectron and the ion.
. By ignoring the residual Coulomb interaction between the photoelectron and the ion (blue curve), the trends seen in Fig. 3(a) can be intuitively understood by comparing the spectral bandwidth of the attosecond pulse with the energy splitting between the ionic states. By including Coulomb interactions within a given channel (green curve), the trends shown in Fig. 3 do not differ qualitatively from the first case, where all Coulomb interactions are ignored. In this second model, the excited electron can only interact with the occupied orbital from which it originates. Interactions between different occupied orbitals are considered only in the full model (red curve). We can clearly see that by including these interchannel coupling terms the degree of coherence differs dramatically for short attosecond pulses (see Fig. 3(a)) and, surprisingly, starts to drop due to an enhanced interaction between the slow photoelectron and the ion.
Our results have far-reaching consequences beyond the atomic case. Molecules will be even more strongly affected by interchannel coupling due to the reduced symmetry and smaller energy splittings between the cation many-electron eigenstates. Interchannel coupling is also likely to be significant for inner-valence hole configurations in molecules, which show strong mixing to configurations outside the TDCIS model space.
The present study suggests that interchannel coupling accompanying the hole creation process will affect attosecond experiments investigating charge transfer processes in photoionized systems. The control of decoherence requires widely tunable attosecond sources, thus offering a new opportunity for x-ray free-electron lasers.
| References | ||||||||||||
|
| Contact information |
|
Robin Santra |
| Further Information |