



")
")

Figure 1: Experimental and computed SAXS scattering data. A, the eRF1/eRF3/GTP complex, B, free eRF1. The logarithm of the scattering intensity is plotted against the momentum transfer s = 4π sinθ/λ, where 2θ is the scattering angle and λ = 1.5 Å is the X-ray wavelength. The plots are displaced along the ordinate for better visualization. (1), experimental scattering; (2) computed scattering from the molecular model of eRF1-eRF3 (in A) and from the crystallographic dimer of eRF1 (in B); (3) computed scattering from these two models after rigid body refinement; (4) computed scattering from the rigid body model of eRF1-eRF3 using extended eRF1 (A) and from the rigid body model of eRF1 dimer using a compact eRF1 monomer (B).
Zhihong Cheng1, Kazuki Saito2, Andrey V. Pisarev3, Miki Wada2, Vera P. Pisareva3, Tatyana V. Pestova3, Michal Gajda4, Adam Round4, Chunguang Kong1, Mengkiat Lim1, Yoshikazu Nakamura2, Dmitri I. Svergun4,5, Koichi Ito2,6, and Haiwei Song1
1. Cancer and Developmental Cell Biology Division, Institute of Molecular and Cell Biology, A*STAR (Agency for Science, Technology and Research), 61 Biopolis Drive, Singapore 138673, Singapore
2. Department of Basic Medical Sciences, Institute of Medical Science, University of Tokyo, 4-6-1 Shirokanedai, Minato-ku, Tokyo 108-8639, Japan.
3. Department of Microbiology and Immunology, SUNY Downstate Medical Center, Brooklyn, NY 11203, USA
4. European Molecular Biology Laboratory, Hamburg Outstation, Notkestrasse 85, 22603 Hamburg, Germany
5. Institute of Crystallography, Russian Academy of Sciences, Leninsky pr. 59, 117333 Moscow, Russia
6. Precursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST),
Published as: “Structural insights into eRF3 and stop codon recognition by eRF1”, Genes and Development Vol. 23 No. 9, 1106-1118, (2009).
Developments in Small-Angle X-ray Scattering (SAXS) at the EMBL, Hamburg Outstation made this method a high-throughput, low resolution tool providing information about overall structure and conformational changes of biological macromolecules in solution. SAXS is often employed along with protein crystallography to elucidate structure of functional complexes, such as that of proteins eRF1 and eRF3, which assure fast release of peptides from the ribosome during termination of protein synthesis. It was found that the M domain of eRF3 stimulates a GTPase activity of eRF1. The latter protein recognizes the stop codon of the messenger RNA, and in concert with eRF3 mediates dissociation of ribosome, which plays a critical role in the recycling of the ribosome before the next translation event.
Synchrotron Small-Angle X-ray Scattering (SAXS) is a rapid technique to study the overall structure of native biological macromolecules in solution. Using modern analysis methods, low resolution shapes of individual macromolecules or complexes may be determined ab initio [1]. For multidomain proteins or complexes, where high resolution structures of domains or subunits are available, rigid body modelling may be employed to determine the quaternary structure [2]. SAXS studies in solution are particularly useful in analyzing changes induced by ligand binding or change of temperature, ionic strength, pH etc. These structural responses may be difficult to assess in a crystal, where the biological macromolecules are constrained by interaction with the crystal lattice. Moreover, the macromolecular complexes may be flexible and/or transient, which makes it difficult to apply high resolution methods like protein crystallography or nuclear magnetic resonance.
The EMBL X33 beamline at DORIS III is optimized for SAXS on bio macromolecular solutions and has recently been upgraded [3] to signifi cantly improve its performance and user friendliness. In particular, the beamline is equipped with a PILATUS photoncounting detector (DECTRIS, Switzerland), and a liquid handling robot for high throughput experiments [4]. An automated data analysis pipeline provides the users with the fully processed data and ab initio three-dimensional structural models within a few minutes following the data collection. The EMBL-developed program package ATSAS [5] allows for further comprehensive structural modelling of the experimental data in terms of biologically sensible models. The X33 beamline is employed in a rapidly growing number of collaborative biological projects by user groups worldwide.
An example of the use of SAXS to validate and complement structural models provided by protein crystallography is given by a recent study of a functional complex involved in termination of protein synthesis in eukaryotes. Termination of protein synthesis occurs when the stop codon of the messenger RNA enters the ribosomal A site, and is conducted in eukaryotes by a complex consisting of release factors 1 (eRF1) and 3 (eRF3). The crystal structure of human eRF1 release factor has been solved [6], and high resolution structures of both yeast eRF1 and eRF3 are also available.
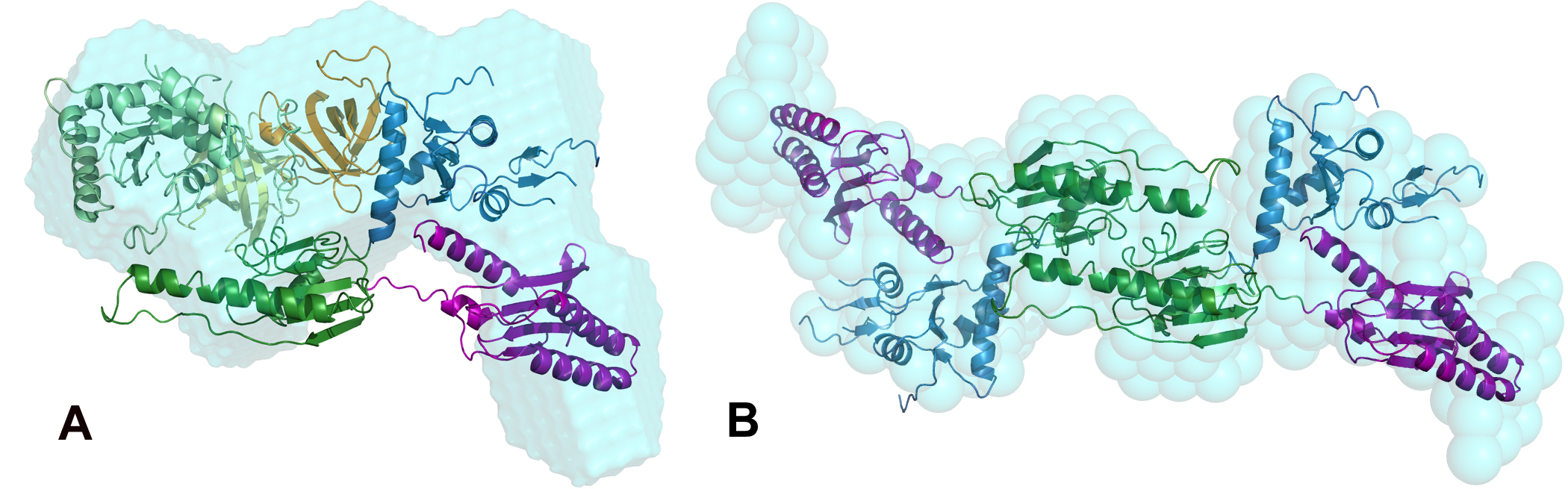
In the work presented here we report on the formation and structure of the full human eRF1*eRF3 complex by crystallography, SAXS, and mutagenesis studies. Crystal structures of full-length eRF1 (human and S. pombe) in complex with a truncated eRF3 (lacking the so-called GTPase domain) were solved. They reveal details of the interface between domain 3 of eRF3 and the domain C of eRF1, and suggest a marked conformational change in eRF1 upon eRF3 binding. SAXS experiments were performed on both components of the complex separately, and on a full complex, see data on Figure 1. These data confirm that compaction of eRF1 occurs only upon binding with eRF3. An ab initio low resolution shape of the complex was computed using DAMMIF [7] (Figure 2a) and revealed structural mimicry to a complex of tRNA with an elongation factor EF-Tu.
The rigid body modeling of the SAXS data from the full-length eRF1*eRF3 complex constrained by the inter-residue contacts between the two proteins in the crystal was done using SASREF [2] to yield a quasi-atomic model in Figure 2a. Here, the C-terminal domains of eRF1 and eRF3 bind together, but also the M domain of eRF1 binds with this eRF3 domain proximal to the hydrolyzation site of guanosine triphosphate, which is known as activator of the peptide release reaction. Biochemical study confirmed the hypothesis, based on SAXS and crystallographic models, that residue Arg192 is stimulating the eRF3 activity in hydrolysing the guanosine triphosphate.
The synergistic study using crystallography, SAXS and mutational analysis provides new structural insights into the stop codon recognition by eRF1 and the process of translation termination in protein synthesis. This underlines the possibilities of SAXS and multidisciplinary approaches for structural characterisation of biomacromolecular complexes.
| References | ||||||||||||||
|
| Contact information |
|
Dmitri I. Svergun |
| Further Information |