


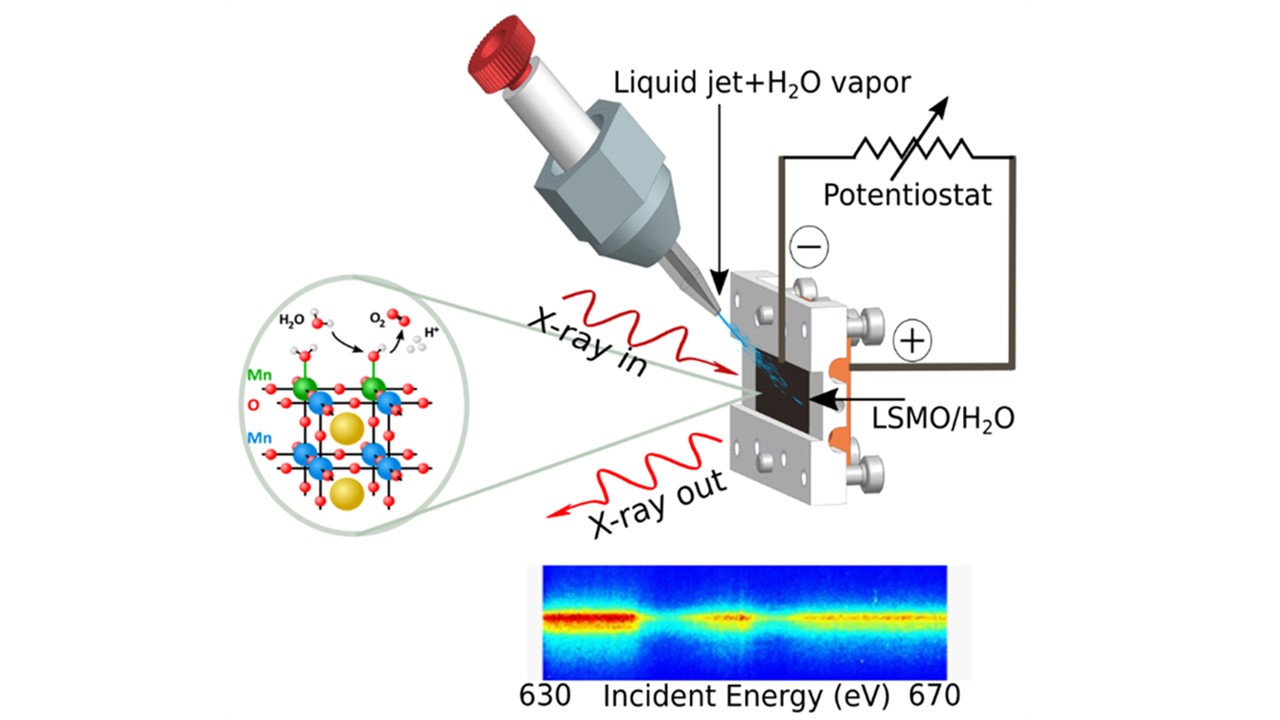
- Our publication on "Atomic Scale Methods in Energy Research" just got pubished in Advanced Energy Materials; DOI: 10.1002/aenm.202404280
- Water splitting: we just came back from our beamtime at Spring-8/Japan

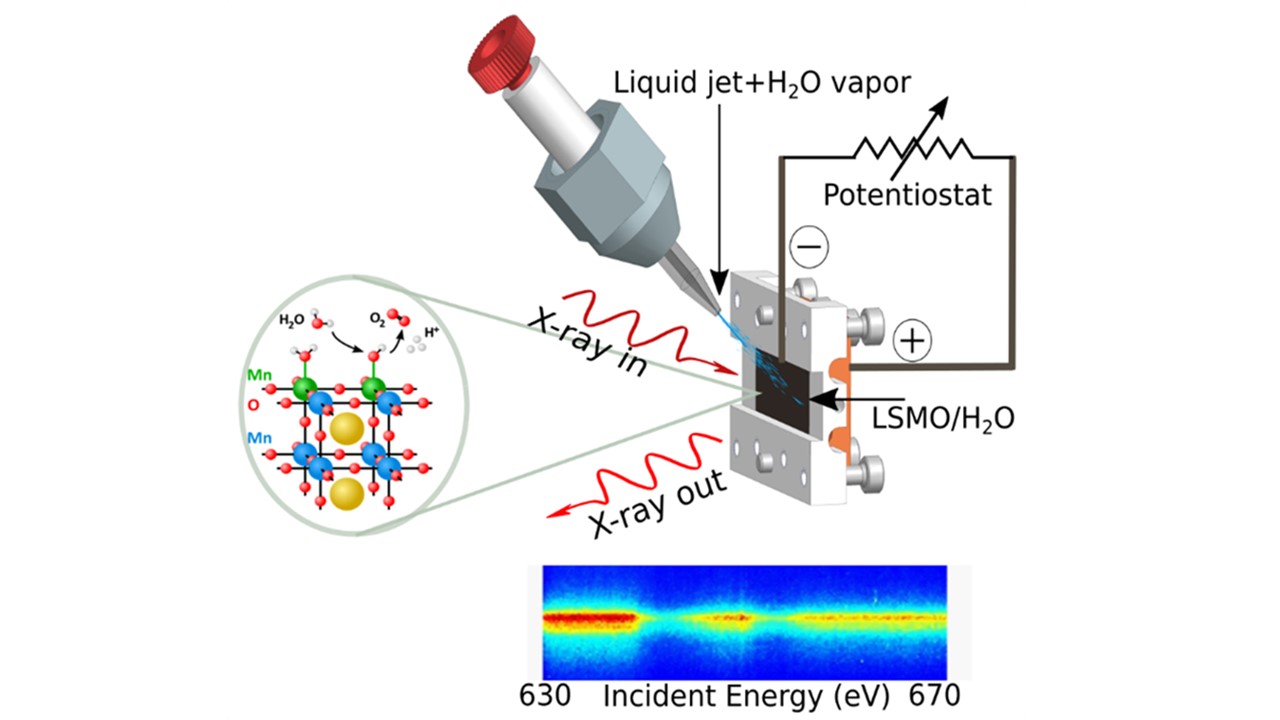

Pulsed X-rays Make us Happy
The website is currently shared between two workgroups – FS-BIG (Prof. S. Bari), concentrating on the study of organic gas phase reactions and the dynamics of biomolecules, and FS-SCS (Prof. S. Techert), focusing on the study of soft matter reactions in the condensed phase (liquid, solid).
The slider object does not work in manage or preview mode.
Structural Dynamics in Chemical Systems
Motivation:
- Exploring the temporal landscape of structural dynamics in soft matter
- We study in particular the mechanisms of complex chemical and biochemical reactions
- Precisely, we study the reacting molecules' time-dependent electronic and spatial behavior during chemical reactions
- For this we employ ultrafast and time-resolved elastic and inelastic X-ray scattering techniques (diffraction and spectroscopy), which we develop for high-flux X-ray sources (synchrotrons and free-electron lasers), the so-called "molecular movie methods" in the time-resolved and ultrafast X-ray field
Aim:
- At a fundamental level, beyond Polanyi, we aim to compress ‘traditional’ or classical reaction coordinates into novel reaction coordinate descriptions that allow us to directly derive, in real time, the changes in physico-chemical parameters from the reactants to the formed ultrafast species / intermediates / metastable states during a reaction (such as the evolution of energy, entropy, energy/entropy flow, structural changes, etc.).
- We thus describe biochemical reactions and function beyond simple “structure-function” principles, and our new insights directly influence the design of novel smart bioorganic and organic materials
History:
- So far, we have studied over 300 different types of chemical and biochemical reactions and found that if we want to understand our “molecular movies” and the reaction’s driving forces, we needed to overcome traditional kinetics descriptions ...
FS-SCS: From Structural to Topological to Functional Dynamics in Chemical Reactions
Responsibles:
Krishnayan Basuroy, Jose Velazquez Garcia, Simone Techert
Method Developments:
Ultrafast X-ray crystallography (ordered system and for high resolution), out of equilibrium crystallography, ultrafast X-ray scattering (disordered systems), pump / probe experiments, sample delivery units (liquid jets for free-electron laser research, nanoliter mixing etc.), combination with ultrafast transient optical spectroscopy, in-situ/operando chemical synthesis and device built-up at high flux X-ray sources (allowing on-the-fly chemical analysis), protein dynamics for artificial applications
Soft Matter Systems and Reactions under Investigation:
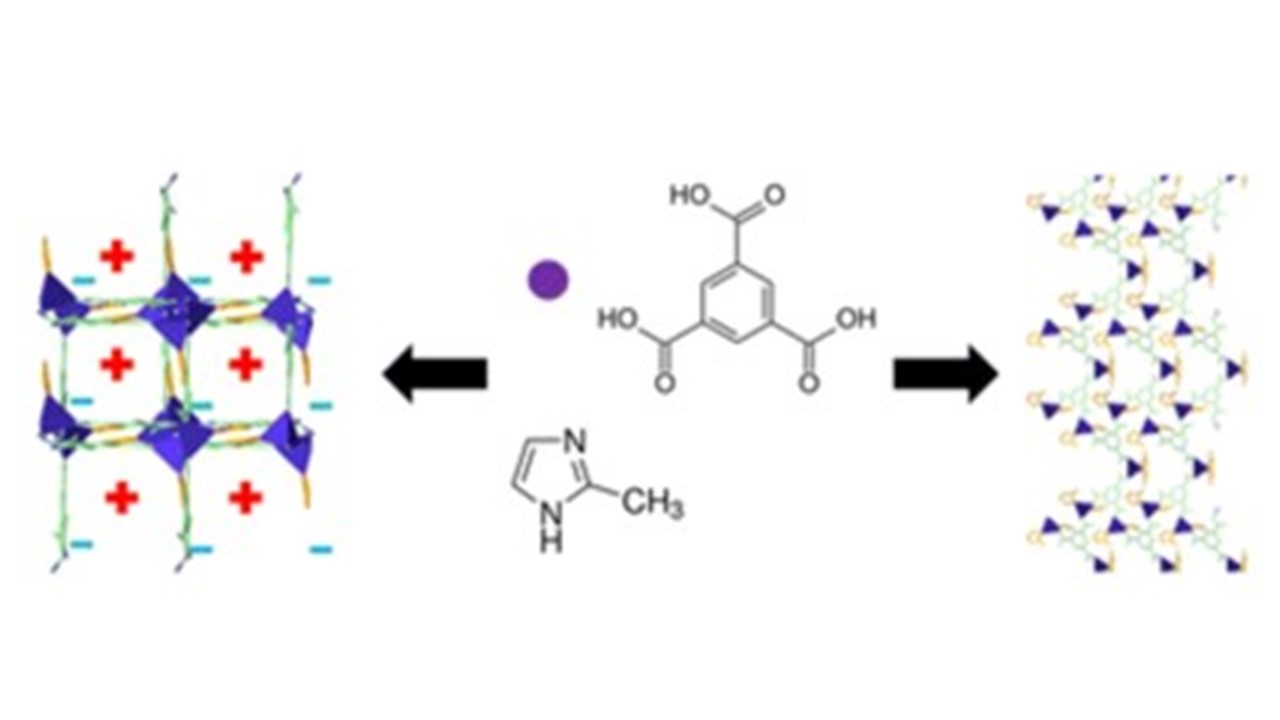
- From electron spins: structural dynamics studies of MOFs, ultrafast spin-crossover talk in iron-grid complexes, investigation of their structure-optical-function relationship on different time-scales (from the ultrafast up to seconds); (JVG+ST)
- to electron orbitals: structural dynamics investigations of molecular electronics: studies on the time-evolution of intra-molecular charge transfer geometries in pyrene-bridge-dimethylaniline dyad crystals, exploration of the dynamics of multi-stimuli responsiveness in dyad crystals (time-resolved / high pressure); (KB+ST)
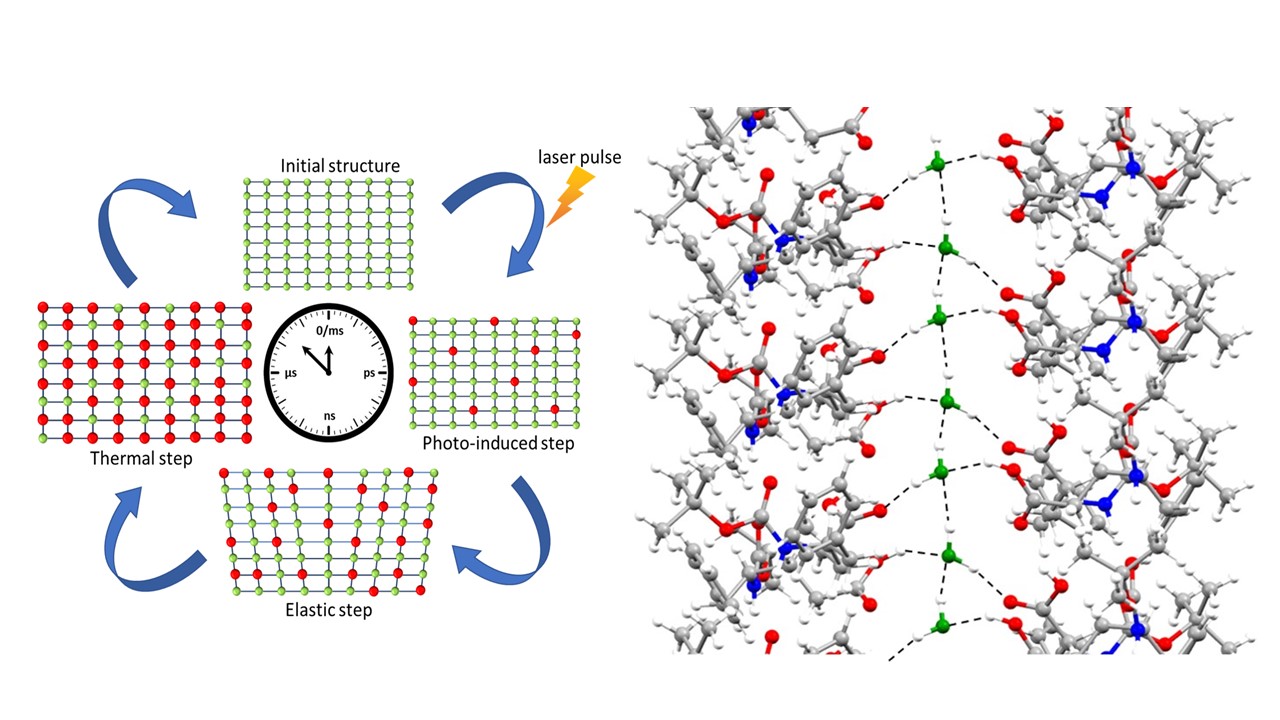
- to hydrogen bridges: structural dynamics exploration of amino-acid / peptide based, self-assembled nano-structures such as nanotubes/ nanochannels/ nanopores (thermally induced); “water nano-zippers”; structural dynamics studies of intrinsically disordered proteins (IDPs), study of hydrogels formed by IDPs, structural dynamics studies of FG containing peptide fragments; structural dynamics of liquid crystal systems (KB+ST)
FS-SCS: From Local to Global Dynamics in Chemical Reactions: Electronic Changes
Responsibles:
Sreeju Sreekantan Nair Lalithambika, Simone Techert
Method Developments:
X-ray photon-in / photon-out techniques, ultrafast multidimensional soft X-ray spectroscopy (including ultrafast soft X-ray absorption spectroscopy, ultrafast soft X-ray emission spectroscopy, ultrafast resonant inelastic X-ray scattering techniques), pump / probe expeirments, liquid-jet endstation (@SXR/LCLS-I), HeisenbergRIXS (@SCS/EuropeanXFEL), in-situ and operando techniques combined with ultrafast pump / probe approaches
Soft Matter Systems and Reactions under Investigation:
- from water splitting reactions: investigations from the catalysts and from the water point of view
- to heterogeneous catalysis: oxygen evolution reaction on perovskites and complex oxide surfaces, dynamics of the Helmholtz layer
- to homogeneous catalysis: hydrogen evolution reaction in homogeneous catalysis, ultrafast dynamics of Rh-catalysts, photo catalysis and solvent influence on the ultrafast time-scale
- to proton-coupled electron transfer reactions: real time monitoring of hydrogen evolution reactions in homogeneous and heterogeneous catalysis
FS-BIG: Structure and Dynamics of Gas-Phase Biomolecules
Responsible: Lucas Schwob, Sadia Bari (please contact Sadia.Bari@desy.de and Lucas.Schwob@desy.de directly)
Method Developments: Ultrafast X-ray Mass Spectrometry, Ultrafast X-ray Circular Dichroic Absorption and Mass Spectrometry, Electrospray Sources for Free Electron Laser Research
Electrospray ionization (ESI) is a gentle, state-of-the-art technique to introduce biomolecular ions from solution into the gas phase and into vacuum, providing a solvent- and substrate-free environment. This allows studying the molecule in a well-defined, isolated state, where only intramolecular interactions have to be considered for describing structure and dynamics. The combination of an ESI source with advanced light sources such as synchrotrons, free-electron lasers and high harmonic generation sources allows for a novel and unique way to investigate structure and dynamics of gas-phase biomolecules. In particular, synchrotron and free-electron laser sources have the great advantage of superior photon brilliance, a wide photon energy range as well as polarization tunability. This enables systematic studies of energy and polarization dependent effects of ionization and dissociation processes.
The following fundamental scientific complexes are targeted within this group:
(i) Circular dichroism: Homochiral molecules have different absorption efficiencies for right or left circularly polarized light, the so-called circular dichroism (CD). Many CD studies aimed at discovering structural information on amino acids, proteins and DNA (building blocks) have been performed in the ultra-violet (UV) spectral region, but only a few CD studies enter the X-ray spectral range, where despite a very localized excitation, strong CD was observed. This local, site-selective probing with an X-ray pulse does not average the signal over the whole molecule, as is the case in the UV-visible absorption, and hence is a more selective tool to investigate structure-dependent dynamics in the molecule.
(ii) Radiation damage: Radiation therapy relies on ionizing radiation, yet, there is no consistent knowledge of the exact cascade of processes leading to radiation damage in a cell on atomic length and time scales. It is important to understand the details in the mechanisms of radiosensitizers, to increase the impact on tumors and to find non-toxic alternatives. To study the ultrafast processes of radiation damage on the molecular level, photoionization and photodissociation experiments are performed on gas-phase nucleobases, amino acids, oligonucleotides and peptides. These molecules will be explored in the pure as well as nanosolvated form. Moreover, metal ligands as well as radiosensitizers like cisplatin attached to oligonucleotides will be studied.
(iii) Ultrafast charge migration: Ultrafast charge transfer is believed to play an important role in biological energy conversion processes, such as light harvesting processes in plants. Pump-probe experiments of peptides will be performed to understand the fast charge migration processes in detail in terms of size, structure and time.
One example of a merge of complementary techniques is the combination of high flux and high energy resolution X-ray spectroscopy with electro spray ionization and mass spectrometry.
Since the absorption of X-ray photons is element specific and selective towards its chemical environment, tuning the X-ray excitations with defined photon properties allows to collect ionization and chemical fragmentation pattern generated in an initial step of “electron time clocking” on this internal femtosecond X-ray clock. Consequently, the chemical fragments are defined through the characteristics of the X-ray excitation. The combination of X-ray spectroscopy with mass spectrometric tools allows for the precise determination of the experimental energies of occupied and low lying non-occupied molecular orbitals of complex molecules (polyaromatic systems, nucleotides,amino acids, peptides, proteins). In comparison, optical spectroscopy in the VIS und UV regime delivers relative transition energies. Therefore it is possible to determine with extremely high precision experimentally binding energies and characterize ionization processes during ion and radical formation or hydrogen bonding processes. The recorded, corresponding decomposition pattern include – again – chemical analytical information typical for mass spectrometry – simply precisely connected to the initial orbitals of biomolecules from which the journey of chemical decomposition starts. Such correlations deliver additional puzzle pieces in determining time stamps of biochemical reactions in structural features of the relevant molecules.
FS-BIG: Ultrafast Imaging of Gas-Phase Reactions
Responsible: Lucas Schwob, Sadia Bari (please contact Sadia.Bari@desy.de and Lucas.Schwob@desy.de directly)
Method Developments: Ultrafast X-ray Photoelectron Spectroscopy, Ultrafast X-ray Photoelectron Imaging, Ultrafast X-ray Coulomb Explosion Imaging, Adds-on to CAMP (FLASH)
Investigating chemical reaction dynamics on the fundamental level, watching the making and breaking of chemical bonds in real time, or recording a “molecular movie” has been a longstanding dream in physical chemistry. Directly imaging the motion of single atoms in space and time is very challenging though, due to the molecular dimensions (~1Å) and the reaction time scales (~1-100fs). The advent of X-ray free-electron lasers that provide very short and very intense X-ray pulses has brought this dream a little closer to reality. Our research activities focus on investigating ultrafast molecular dynamics of gas-phase molecules, studied at accelerator-based lightsources.
Molecular physics and photochemistry are omnipresent in everyday life: light-emitting diodes, photovoltaics, biosynthesis of vitamin D, UV-resistance of DNA, and many other processes are based on photochemical principles. However, some of the very fundamental reactions such as electrocyclic ring opening are not yet fully understood on the atomic level. If we can gain the necessary insight to understand, and eventually maybe even to control the outcome of such photochemical reactions, this would open the door to develop more efficient solar cells, to highly innovative medical applications, and many more. The investigation of such ultrafast processes in molecules with optical spectroscopy is referred to as femtochemistry, for which the Nobel Prize in chemistry was awarded to Ahmed Zewail in 1999.
Directly imaging the motion of single atoms in space and time is very challenging though, due to the molecular dimensions and the reaction time scales. The typical bond distance between two atoms in a molecule is on the order of 1 Ångström (1 Å = 10-10 m) and rearrangement of atoms can happen on a time scale faster than 100 femtoseconds (1fs = 10-15 s). Our goal is to develop methods capable of imaging ultrafast processes on the atomic length and time scales in isolated molecules, in particular using ion momentum and electron spectroscopy following X-ray absorption. We approach this by starting with isolated atoms and then gradually turn towards larger molecules. Our main focus lies on time-resolved experiments employing electron and or ion momentum spectroscopy (figure right side).
The advent of X-ray free-electron lasers such as FLASH and the European XFEL at DESY in Hamburg, the LCLS in California, and SACLA in Japan, brought forward unprecedented possibilities to study dynamical processes in the gas phase. The very short and very intense X-ray pulses made it possible, for the first time, to probe ultrafast photo-induced molecular dynamics following multiple, element-specific inner-shell absorption. Such X-ray pulses can remove many electrons from one specific atom in a larger molecule, thus generating a very high, localized charge. This charge can then spread to the molecular environment within a very short time. Charge transfer mechanisms play an important role in a variety of systems, and they lie at the heart of radiation damage processes that have been shown to occur in bioimaging experiments employing very intense X-ray radiation. Even in astrophysics, X-rays produced by charge transfer processes have been observed.
In order to record a “movie” of the molecular dynamics, the same measurement has to be repeated many times, yielding different snapshots of the atomic motion; this is called a pump-probe experiment. Through photoabsorption, it is possible to promote a molecule to a specific excited state. For example, the schematic potential energy curves (PECs) of fluoromethane (CH3F) and iodomethane (CH3I) illustrate that the different halogen species give rise to qualitatively different PECs. Absorption of one ultraviolet photon in CH3I leads to a repulsive neutral state, whereas multi-photon UV absorption in CH3F populates several higher-lying ionic states. After a tunable time delay, an intense X-ray pulse probes the dissociating system by ionizing predominantly the iodine (3d) or the fluorine (1s) level, respectively, because of their large absorption cross sections, resulting in a localized positive charge on the halogen. If the halogen atom is already ionized, the high charge stays on this atom and the rest of the molecule remains neutral. As long as the two fragments are close to each other, the charge can spread over to the other atoms. It is this transition from a bound molecule to isolated atoms that we probe by time-resolved ion spectroscopy. The recorded ions can be used to extract the critical internuclear distance, up to which electron transfer from methyl to iodine is possible. We have observed signatures of long-distance intramolecular electron transfer (up to 15 Å for I21+), which is, among others, highly relevant for radiation damage in imaging experiments involving heavy atomic species.
References
Selected publications
S. S. Nair Lalithambika, ST et al., Advancing Energy Materials by In-situ Atomic Scale Methods, Adv. En. Mat. (March 2025). DOI: 10.1002/aenm202404280
J. Schlappa, SSNL, TR, ST et al., The Heisenberg-RIXS Instrument at the European XFEL, J. Syn. Rad. 32, 29-45 (2025). DOI: 10.1107/S1600577524010890
J. J. Velazquez-Garcia, KB, ST et al., Out-of-Equilibrium Dynamics of a Grid-Like Fe(II) Spin Crossover Dimer Triggered by a Two-Photon Excitation, Chem. Sci. 15, 13531-13540 (2024). DOI: 10.1039/d4sc02933j
K. Basuroy, JJVG, ST et al., Investigation of Encapsulated Water Wire within Self-assembled Hydrophilic Nanochannels, In a Modified 4-Amino Acid Crystals: Tracking Thermally Induced Changes of Intermolecular Interactions within a Crystalline Hydrate, Amino Acids 56, 9 (2024). DOI: 10.1007/s00726-023-03372-4
T. Reuss, SSNL, ST et al., Advancements in Liquid Jet Technology and X-ray Spectroscopy for Understanding Energy Conversion Materials during Operation, Acc. Chem. Res. 56, 203-214 (2023). DOI: 10.1021/acs.accounts.2c00525
K. Schubert, ST, LS, SB et al., Unravelling the Electronic Structure and Deexcitation Pathways of Isolated Cobalt Protoporphyrin IX at the Cobalt L-edge, Chem. Sci. 12, 3966-3976 (2021). DOI: 10.1039/D0SC06591A
S. Bari, ST et al., High flux X-ray sources and Free Electron Lasers for studying ultrafast time structure imprints in complex chemical and biochemical reactions, in: X-ray Free Electron Lasers, eds. U. Bergmann, P. Pellegrini, Oxford University Press (2016). DOI: 10.1039/9781782624097
ST, F. Schotte, M. Wulff, Picosecond x-ray diffraction probed transient structural changes in organic solids, Phys. Rev. Lett. 86, 2030-2034 (2001). DOI: 10.1103/PhysRevLett.86.2030
Our Group Members Work Interdisciplinary
Our group members are chemists, physicist, crystallographer or molecular biologist. Our lively discussions form an inspiring, stimulating and creative room for ideas between the boarders of the scientific disciplines, and allow for the development of novel scientific ideas and solutions in-between. We develop instrumentation for high flux X-ray sources which are necessary for our structural dynamics studies on complex chemical and biochemical reactions and systems, but we also try to understand our studied reactions from first principles in chemical reaction dynamics.
The entire content of the FS-SCS website is protected by copyright (including scientific terminology). Please quote accordingly, if you use content or terms from these pages. © DESY Photon Science