


Figure 1: Molecular structures with atom numbering scheme in the reactive region of aziridine I, oxirane II and olefin III.

Simon Grabowsky1, Thomas Pfeuffer2, Wolfgang Morgenroth3,4,5, Carsten Paulmann3,6, Tanja Schirmeister2, Peter Luger1
1 Freie Universität Berlin, Institut für Chemie und Biochemie / Kristallographie, Fabeckstr. 36a, 14195 Berlin, Germany
2 Universität Würzburg, Institut für Pharmazie und Lebensmittelchemie, Am Hubland, 97074 Würzburg, Germany
3 c/o DESY/HASYLAB, Notkestr. 85, 22607 Hamburg, Germany
4 Georg-August-Universität, Institut für Anorganische Chemie, Tammannstr. 4, 37077 Göttingen, Germany
5 Aarhus University, Department of Chemistry, Langelandsgade 140, 8000 Aarhus C, Denmark
6 Universität Hamburg, Mineralogisch-Petrographisches Institut, Grindelallee 48, 20146 Hamburg, Germany
Published as: “A Comparative Study on the Experimentally Derived Electron Densities of three Protease Inhibitor Model Compounds”, Org. Biomol. Chem. 6, 2295-2307 (2008).
In order to contribute to a rational drug design with ground-state electron densities, it is important to evaluate the deduced functions and properties that provide information on reactivity. Electrostatic potentials mapped on molecular surfaces and Zero-Laplacian iso-surfaces can be referred to as reactive surfaces because they show centres of nucleophilicity or electrophilicity. Derived from experimental densities, these surfaces are directly dependent on intermolecular interaction patterns within the crystal environment. Therefore, the information deduced from experimental electron densities are generally a better approximation for biological enzyme-ligand interactions than gas phase calculations or properties derived from the liquid phase. Within this study, we have determined the electron densities of three protease inhibitor model compounds: acceptor substituted aziridine, oxirane and olefin.

Single crystals of aziridine I and olefin III (see Figure 1) were measured at beamline D3 of HASYLAB at 9K. For aziridine I, 163095 reflections were collected of which 18527 were unique with a completeness of 84.5% to a resolution of 1.02 Å-1. For olefin III, 104810 reflections were collected of which 9203 were unique with a completeness of 90.2% to a resolution of 1.02 Å-1. A single crystal of oxirane II was measured at beamline F1 of HASYLAB at 100 K. 126224 reflections were collected of which 14724 were unique with a completeness of 84.2% to a resolution of 1.19 Å-1. All compounds crystallized in the space group P1. For integration, the program XDS [1] was used and oblique incidence correction [2] was applied. The electron density distribution obtained from multipole refinement [3] with XDLSM of the XD2006 suite of programs [4] was analysed with XDPROP and MolIso [5].
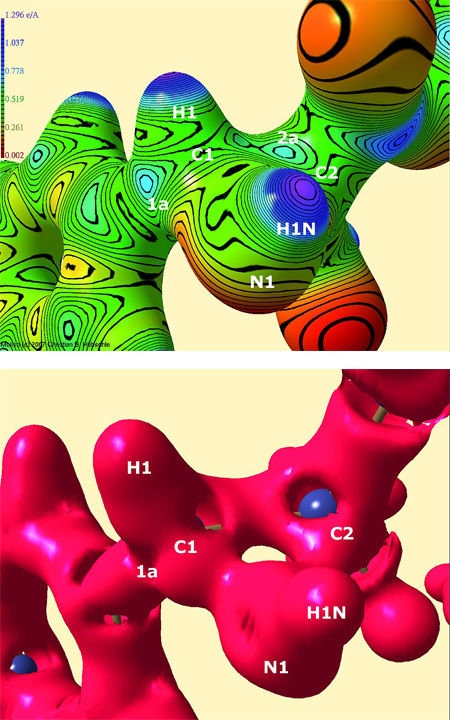
Figure 2: Top: Electrostatic potential in e Å-1 of the reactive region of aziridine I, mapped on an isosurface of the electron density at 0.5 e Å-3.
Bottom: Zero-Laplacian iso-surface of the reactive region of aziridine I.
Positions of atoms under the surface and maxima of the electrostatic potential or reduced valence shell charge concentrations in the Zero-Laplacian at C1 and C2 are plotted.
To correlate the biological activity with the electrostatic potential (esp), it has become common practise to scrutinize the electrostatic potential on a molecular surface of 0.001 a.u. (=0.0067 e Å-3) [6]. To quantify the effects of the esp on this surface, the average deviation Π from the mean surface potential can be used. It is a measure of the internal charge separation or local polarity which is present even in molecules having zero dipole moment. For compounds I to III the values of Π are: Π(cpd. I)=0.089 e Å-1, Π(cpd. II)=0.035 e Å-1 and Π(cpd. III)=0.079 e Å-1. These differences in the overall polarization are caused by the different intermolecular interactions and the structural differences in the reactive regions. The intermolecular bonding network for aziridine I is stronger than for the other compounds and is therefore responsible for the greater overall polarization. These findings may contribute to an explanation for the greater reactivity of aziridine I in biochemical model reactions compared to oxirane II or olefin III.
To obtain information about the location of possible electrophilic centres that can be target of a nucleophilic attack followed by the formation of a covalent bond (ring-opening reaction), an electrostatic potential mapped on a density surface that is much closer to the nuclei should be examined. Figure 2 (top) shows the electrostatic potential of aziridine I mapped on the density isosurface of 0.5 e Å-3. Several maxima (green/blue colour) in the reactive region can be found on the isosurface (most important ones labeled 1a and 2a in Figure 2). The maximum labeled 1a seems to be the sterically most favorable reaction site because there is no bulky sidechain and no negatively polarized carbonyl oxygen atom nearby as it is in the case of the maxima around C2. This finding is confirmed by biochemical model reactions: Carbon atom C1 is attacked regiospecifically. All the other maxima at C1 cannot be reached from the direction of the flat aryl plane but only from the side of the aryl group where hydrogen or nitrogen atoms hinder the attack. Moreover, the maximum 1a has the highest esp value compared to all the maxima around C1 (esp(1a)=0.73 e Å-1). So even stereoselectivity of the ring-opening reaction can be predicted.
The Zero-Laplacian iso-surface (defined by  shows the location of electrophilic centres by means of reduced valence shell charge concentrations (reduced VSCCs) which appear as holes in the surface. Figure 2 (bottom) shows the isosurface of aziridine I. There are larger reduced VSCCs at carbon atom C1 (four large holes at C1, two large and one small hole at C2) so that the experimentally found regioselectivity of the ring opening reaction can again be confirmed. Another important information from the Zero-Laplacian iso-surface are the positions of the reduced VSCCs around the atoms: Although their number and the number of the maxima in the esp do not agree, the agreement of the positions is quite high. Exactly on the position where the maximum in the esp labeled 1a was found, a reduced VSCC can be found in Figure 2 (bottom), too. This is a further hint that this sterically most favorable position is also electronically preferred.
shows the location of electrophilic centres by means of reduced valence shell charge concentrations (reduced VSCCs) which appear as holes in the surface. Figure 2 (bottom) shows the isosurface of aziridine I. There are larger reduced VSCCs at carbon atom C1 (four large holes at C1, two large and one small hole at C2) so that the experimentally found regioselectivity of the ring opening reaction can again be confirmed. Another important information from the Zero-Laplacian iso-surface are the positions of the reduced VSCCs around the atoms: Although their number and the number of the maxima in the esp do not agree, the agreement of the positions is quite high. Exactly on the position where the maximum in the esp labeled 1a was found, a reduced VSCC can be found in Figure 2 (bottom), too. This is a further hint that this sterically most favorable position is also electronically preferred.
|
References |
||||||||||||
|
||||||||||||
|
Contact information |
||||||||||||
|
Peter Luger |
||||||||||||
| Further Information |